Angelmanův syndrom
Z WikiSkript

| Angelmanův syndrom | |
.png) pětiletá dívka z Mexika s Angelmanovým syndromem | |
| Klinický obraz | málo rozvinutá řeč, těžká mentální retardace, motorické problémy, bezdůvodné záchvaty smíchu, hypotonie, mikrocephalie, abnormální EEG |
|---|---|
| Příčina | delece v úseku 15q11–13 na maternálním chromosomu |
| Diagnostika | postnatální (karyotyp, klinický obraz) |
| Incidence ve světě | 1/16 000 narozených |
| Prognóza | lék neexistuje, jedinci potřebují pomoc s základnými činnostmi, doživají se průměrného věku, klinický obraz se věkem mění (může se zmírňovat) |
| Klasifikace a odkazy | |
| MKN-10 | Q93.5 |
| MeSH ID | D017204 |
| OMIM | 105830 |
| orphanet | ORPHA72 |
| MedlinePlus | 007616 |

Angelmanův syndrom (AS, Happy puppet syndrome) je mikrodeleční syndrom, způsobený nejčastěji delecí v úseku 15q11–13 na maternálním chromosomu (tzn. určitého úseku na dlouhém raménku 15. chromosomu) nebo uniparetální dizomií otcovského 15. chromosomu.
Incidence toho syndromu je asi 1/16 000.
Etiologie[upravit | editovat zdroj]
Etiologie Angelmanova syndromu úzce souvisí se syndromem Prader-Willi. Oba syndromy mají rozlišný fenotyp, ale úsek delece je shodný u obou. O tom, kterým z nich bude jedinec postižený, tak rozhoduje genomový imprinting.
70% mikrodelece chromozomu (15q11–13)mat[upravit | editovat zdroj]
- V deletovaném úseku se vyskytují dva kritické genové úseky, které označujeme :
- PWCR
- ASCR
- Angelmanův syndrom potom vzniká tak, že na maternálním patnáctém chromosomu dochází sice k deleci obou úseků, ale důsledkem toho je na otcovském homologním chromosomu methylován úsek ASCR, čímž dojde k jeho inaktivaci. Zůstává tedy aktivní pouze úsek PWCR na otcovském chromosomu.
- Pro úplnost syndrom Prader Willi vzniká naopak tím, že delece nastala na otcovském 15. chromosomu, a tím pádem byl inaktivován úsek ASCR na maternálním.
Uniparentální dizomie otcovského nebo mateřského chromozomu[upravit | editovat zdroj]
- V některých případech, kdy u postiženého nelze prokázat deleci 15q11–13 je syndrom pravděpodobně způsoben uniparentální dizomií. To znamená, že jedinec dostal oba chromosomy 15 od jednoho rodiče a má tedy dvě identické kopie jednoho chromosomu od jednoho z rodičů. To od kterého z rodičů chromosomy zdědil opět ovlivňuje projev příslušného syndromu. Tedy pokud :
- má oba chromosomy od matky – vzniká Prader-Willi syndrom, a dochází k inaktivaci obou úseků PWCR
- má oba chromosomy od otce – vzniká Angelmanův syndrom a oba úseky ASCR jsou methylovány
Jiné mutace[upravit | editovat zdroj]
- speciálními molekulárně biologickými vyšetřeními byly zjištěny i jiné příčiny vzniku Angelmanova syndromu jako například
- mutace genu E6–AP (UBE3A), který kóduje ubikvitin-protein ligázu nebo
- u 7–9 % pacientů postižených Angelmanovým syndromem byly prokázány mutace imprintingového centra, řídícího imprinting ostatních genů a nacházejícího se právě v kritickém úseku.
Klinický obraz[upravit | editovat zdroj]
Syndrom se častěji vyskytuje u dívek, ale není to pravidlem. Děti potřebují celoživotní asistenci a asi 70 % z nich se dokáže naučit vykonávat jednoduché domácí práce.
Fenotyp Angelmanova syndromu[upravit | editovat zdroj]
- u všech postižených pozorujeme :
- málo rozvinutou řeč – pouze minimum slov, spíše neverbální projev
- těžká mentální retardace v pásmu debility až imbecility
- motorické problémy – ataktické pohyby, strnulá chůze (připomínající pohyby loutky)
- bezdůvodné záchvaty smíchu
- asi u 80 % se vyskytuje:
- porucha pozornosti
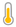 hypotonie
hypotonie- mikrocephalie
- abnormální EEG
- u 20–80 % se může objevit i :
- šilhání
- poruchy polykacího reflexu
- hypopigmentace
- oploštělé záhlaví
- hyperaktivita
- epileptické záchvaty
Odkazy[upravit | editovat zdroj]
Související články[upravit | editovat zdroj]
- Genový imprinting a lidské patologie
- Genový imprinting
- Beckwithův-Wiedemannův syndrom
- Prader-Williho syndrom
- Uniparentální disomie
Externí odkazy[upravit | editovat zdroj]
Zdroj[upravit | editovat zdroj]
- KOČÁREK, Eduard, Martin PÁNEK a Drahuše NOVOTNÁ. Klinická cytogenetika I.: úvod do klinické cytogenetiky, vyšetřovací metody v klinické cytogenetice. 1. vydání. Praha : Karolinum, 0000. 120 s. ISBN 80-246-1069-8.
- ŠTEFÁNEK, Jiří. Medicína, nemoci, studium na 1. LF UK [online]. [cit. 11. 2. 2010]. <https://www.stefajir.cz/>.