Neuroblastom


| Neuroblastom | |
| 9500/3 | |
 CT snímek zachycující neuroblastom v játrech | |
| Lokalizace | extrakraniální |
|---|---|
| Incidence v ČR | 7,5:100000 kojenců |
| Prognóza | závislá na stadiu, při včasné diagnostice 95–100% přežití |
| Klíčová mutace | zvrat buněk truncus sympathicus |
Neuroblastom je embryonální maligní nádor raného dětského věku, vycházející z buněk nervové tkáně. Jedná se o nádor solidní s extrakraniální lokalizací. Vzniká maligním zvratem nezralých buněk sympatiku, které pocházejí z neurální lišty. Je typický svým variabilním chováním – spontánní regrese (více než 10 % nádorů), vyzrávání v benigní ganglioneurom (méně časté), ale i výskyt vysoce maligních forem s rozsevem metastáz. Primární nádor může být lokalizován v dutině břišní (celkem 65 % − nadledvina 40 % a retroperitoneum 25 %), v mediastinu (15 %), v krční oblasti (5 %) a v pánevních sympatických gangliích (5 %). Přibližně v 1 % případů se nepodaří lokalizaci primárního nádoru určit. Neuroblastom metastazuje lymfatickou i krevní cestou, metastázy zakládá v lymfatických uzlinách, kostní dřeni, kortikální kosti, dura mater, orbitě, játrech a kůži. Méně častěji umí metastazovat také do plic či intrakraniálně.
Epidemiologie[upravit | editovat zdroj]
V České republice je neuroblastom diagnostikován přibližně u 20–30 dětí ročně. U kojenců jde o nejčastější maligní nádorové onemocnění vůbec. 90 % všech neuroblastomů je diagnostikováno do 5 let věku dítěte. Výskyt u adolescentů je vzácný, u mladých dospělých raritní.
Histologický obraz[upravit | editovat zdroj]
Mikroskopicky se jedná o nádor z malých modrých kulatých buněk dětského věku. Jádro je hyperchromatické, lem cytoplazmy úzký, nádorové buňky mají tendenci k tvorbě rozet. Na základě histologie lze rozlišit prognosticky příznivé a nepříznivé typy nádoru.
Klinický obraz[upravit | editovat zdroj]
Časté jsou nespecifické celkové příznaky:
- únava a slabost,
- nechutenství,
- úbytek hmotnosti,
- neprospívání,
- změny chování,
- zvýšené teploty,
- anémie,
- otoky,
- hypertenze.
Další příznaky vycházejí z lokalizace nádoru (či metastáz):
- dutina břišní − hmatná rezistence či rovnou deformace břišní stěny, bolesti břicha, anorexie, obstipace,
- hlava a krk − exoftalmus, Hornerův syndrom, strabismus, edém papily, atrofie optiku, unilaterální obstrukce nosní dutiny,
- intraspinální lokalizaci− bolesti v zádech, poruchy čití, motorické defekty až paraplegie,
- mediastinum − dyspnoe, dysfagie, opakované infekce dýchacích cest,
- pánevní lokalizace − poruchy mikce a/nebo defekace,
- kožní metastázy − rezistence fialového vzhledu,
- kostní metastázy − bolesti kostí, odmítání chůze a kulhání,
- metastázy v orbitě − hematomy víček, protruze a deviace bulbů.
Méně časté, ale typické projevy:
- akutní encefalopatie mozečku,
- paréza dolních končetin − intraspinálního šíření nádoru (primárně paraspinální nádor),
- těžký průjem − produkce vazoaktivního intestinálního pepitu,
- Hornerův syndrom − postížení sympatických ganglií krku a horní části hrudníku,
- hypertenze s pocením a zčervenáním − produkce katecholaminů.
Diagnostika[upravit | editovat zdroj]
Přibližně 90 % neuroblastomů u dětí je diagnostikováno v prvních 5 letech života. Vychází z fyzikálního vyšetření, zobrazovacích metod, laboratorního vyšetření a chirurgické biopsie. Zcela zásadní je diagnostika histopatologická s klasifikací tumoru pro stanovení rizikové skupiny, prognózy a léčebného schématu.
Klasifikace[upravit | editovat zdroj]
Klasifikuje se do klinických stádií dle mezinárodně platného doporučení (International Neuroblastoma Staging System − INSS) do 4 stupňů: I., II.A, II.B, III., IV., IV.S. Dále se používá nová mezinárodní klasifikace (International Neuroblastoma Risk Groups − INRG), která je založena na věku pacienta (≤18 a >18 měsíců), stadiu onemocnění (lokalizované vs. metastatické), genetických změnách (stav protoonkogenu MYCN) a na přítomnosti rizikových faktorů chirurgických a zobrazovacích metod. Dle rizikových faktorů se onemocnění dělí na nízké, střední a vysoké riziko.
Diferenciální diagnostika[upravit | editovat zdroj]
V úvahu musíme brát Wilmsův tumor, ostatní germinální nádory, dále pak non-Hodkingské lymfomy, Ewingův sarkom, rhabdomyosarkom a další nádorová onemocnění dětského věku. Nelze zapomenout ani na nenádorové příčiny – např. cysty.
Léčba[upravit | editovat zdroj]
Většina lokalizovaných forem I. a II. stadia se léčí pouze chirurgicky, přičemž některé spontánně regredují a operace tak není nutná. Terapie nádorů III. stadia kombinuje operaci a chemoterapii. U nejrizikovějších forem je do terapie zařazována megachemoterapie s následnou autologní transplantací kostní dřeně.
Prognóza[upravit | editovat zdroj]
Je závislá na stupni rizika, odvozeném ze stupně malignity nádoru. U nízkého stupně malignity je tříleté přežití 95–100 %, u středního 85–90 %, u vysokého stupně rizika klesá pod 35 %. Věk pod 1 rok se považuje za pozitivní prognostický faktor, protože u těchto pacientů je vetší šance, že nádor bude vyzrávat. Přibližně 40 % pacientů má v době diagnózy generalizovanou formu nemoci s metastázami.

Neuroblastom − drobné buňky uspořádané do rozet.
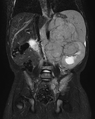
MRI snímek – neuroblastom v retroperitoneu u 2leté pacientky.

Dítě s hepatomegalií a deformací břišní stěny způsobenou metastázami neuroblastomu.



Odkazy[upravit | editovat zdroj]
Související články[upravit | editovat zdroj]
Použitá literatura[upravit | editovat zdroj]
- ŠTĚRBA, Jaroslav, Pavel MAZÁNEK a Viera BAJČIOVÁ. Pokroky v diagnostice a léčbě neuroblastomu u dětí [online]. [cit. 2012-01-07]. <https://zdravi.euro.cz/clanek/postgradualni-medicina/pokroky-v-diagnostice-a-lecbe-neuroblastomu-u-deti-162702>.
- ŠNAJDAUF, Jiří a Richard ŠKÁBA. Dětská chirurgie. 1. vydání. Praha : Galén, 2005. ISBN 807262329X.
- LEBL, Jan, Jan JANDA a Jan STARÝ, et al. Klinická pediatrie. 1. vydání. Praha : Galén, 2012. 698 s. ISBN 9788072627721.