Portál:Otázky z patologie (1. LF UK, VL)/17. Otázka

 | 17. Otázka | |||
| Intracelulární akumulace lipidů – steatóza, lipidózy | ||||
| Otázky z patologie (1. LF UK, VL) | ||||
| ⏪️ ⏩️ | ||||
Steatóza
Steatóza je abnormální hromadění lipidů v buňkách parenchymových orgánů. Vzniká poruchou metabolismu s intracelulární akumulací abnormálního množství lipidů.
Projevy steatózy
Mikroskopicky můžeme v buňce vidět tukové kapénky volně v cytoplazmě nebo obalené membránou. Kapének může být přítomno mnoho (malokapénková steatóza) nebo jedna velká kapénka (velkokapénková steatóza), která utlačuje organely buňky na periferii.
Makroskopicky bývají orgány žluté (prokrvené až do oranžova), na řezu mastné. Nejčastěji jsou postiženy buňky parenchymatických orgánů – játra, ledviny, srdce a kosterní svaly. Příčinami vzniku steatózy jsou především obezita, alkohol, diabetes mellitus, ale také toxiny, hypoxie a proteinkalorická malnutrice.
Dělení steatózy dle mechanismu vzniku
Steatóza může vzniknout poruchou funkce některých buněčných organel – endoplazmatického retikula, mitochondrií nebo lysozomů.
Porucha syntézy a sekrece lipoproteinů
Porucha endoplazmatického retikula, které není schopno produkovat apoprotein B nebo MTP (protein přenášející triacylglyceroly). Postihuje tedy buňky, které lipoproteiny vytváří – hepatocyty (VLDL, Apo-B-100) a enterocyty (chylomikra, Apo-B-48). Porucha endoplazmatického retikula může být buďto dědičná nebo získaná (např. při otravě CCl4).
Steatózu může způsobit hladovění, především u diabetiků. Nedostatečná utilizace glukózy způsobí zvýšenou mobilizaci mastných kyselin. Tělo diabetika není schopno vytvořit dostatečné množství lipoproteinů, což způsobí hromadění mastných kyselin.

Mitochondriální insuficience
Steatóza nastává tehdy, kdy se neutilizované MK v cytoplasmě reesterifikují na TAG. Ty pak tvoří v cytoplasmě tukové kapénky, které jsou vázány na mitochondrie.
K mitochondriální insuficienci dochází při poruše karnitinového přenašeče, kdy se mastná kyselina nemůže dostat do mitochondrie. Porucha karnitinového přenašeče může být dědičná (vrozený deficit karnitinu) nebo získaná (difterotoxin).
Další příčinou snížené utilizace mastných kyselin je porucha beta-oxidace v mitochondriích. Nejčastějším důvodem je ischemie a hypoxie, toxické vlivy (alkohol), případně dědičné choroby. Dochází k ní v energeticky náročných buňkách – kosterní svalovina, myokard, hepatocyty, buňky renálních tubulů.
V játrech nastává steatóza například při hladovění. Jak již bylo zmíněno, dojde ke zvýšené mobilizaci mastných kyselin. Mitochondrie hepatocytů nejsou schopny dostatečně rychle mobilizovat mastné kyseliny a zároveň není dostatečná tvorba lipoproteinů. Játra se steatózou mají typický makroskopický vzhled a nazývají se muškátová játra (na řezu připomínají muškátový oříšek – centroacinózní steatóza).
Pro myokard je typická hypoxická steatóza myokardu (především při těžkých anémiích). Tuk se ukládá podél venózních úseků kapilární sítě, což způsobí typické žlutavé žíhání srdce – tygří (nebo drozdí) srdce.
Defektem β-oxidace (deficit dehydrogenázy) je také vysvětlován Reyův syndrom, při němž často dochází k náhlým úmrtím dětí (např. při respirační infekci dojde k uvolnění MK z tukové tkáně a z důvodu mitochondriální insuficience k nakupení MK a jejich derivátů zvláště v játrech (masivní steatóza – akutní jaterní selhání) a mozku (mozkový edém).
Lysozomální dysfunkce
Zvýšená endocytóza lipidů
Závisí na funkci histiocytů (lipofágy nebo pěnité buňky) s intenzivní endocytózou lipoproteinů nebo jiných struktur bohatých na lipidy (krevní destičky, erytrocyty, myelin). Lipidy se nejprve deponují intralyzosomálně (kapénky obalené membránou), později v cytoplasmě.
Většinou jde o cholesterol, který je v cytoplasmě esterifikován a poté odstraňován HDL. Sekundárně se může zmnožit lipopigment ceroid. Pěnité buňky nacházíme v malatických oblastech mozku, ve sliznici žlučníku (cholesterolóza – jahodový žlučník), v chronických abscesech, aterosklerotických plátech, xantomech nebo v žaludeční sliznici.
Lipidózy
Lipidózy jsou steatózy z důvodu vrozené poruchy enzymů lipidového metabolismu. Jde především o lysozomální hydrolázy, které působí rozkládání složených lipidů (hromadění lipidů v lysozomálním aparátu). Následkem toho lysozomální aparát hypertrofuje a získává typický voštinovitý tvar.
-

Steatoza jater v histologickém preparátu -
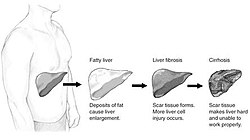
Průběh poškození jater počínající steatózou -

Steatóza na CT



Hyperlipémie
Heterogenní skupina metabolických onemocnění, kdy dochází ke zvýšené hladině lipidů a lipoproteinů v plazmě. Můžeme ji považovat za extracelulární steatózu.
LDL částice jsou v těle transportovány do hepatocytů, ale také do monocytů, které mají receptor i pro chemicky poškozené LDL. Při hypercholesterolémii se transport LDL do histiocytů a do cévní stěny zvyšuje (menší molekuly snáze projdou endotelem). Další příčinou může být zvýšená syntéza lipoproteinů, chybění enzymu, který hydrolyzuje transportovaný lipid (na povrchu endotelu či hepatocytů), nebo chybění receptorů pro LDL na periferních buňkách. Zvýšená hladina lipoproteinů v krvi může urychlovat aterosklerózu.

Hyperlipémie mohou být buďto primární – geneticky podmíněné nebo sekundární, kdy doprovází jiná onemocnění.
Familiární hypercholesterolémie (primární HLP)
Mutace genu pro LDL receptor (transport a metabolismus cholesterolu), která způsobí zvýšenou hladinu plazmatického cholesterolu. Jedinci postižení touto chorobou mívají po narození třikrát (i více) zvýšenou hladinu cholesterolu.
Důsledkem tohoto onemocnění je předčasná ateroskleróza koronárních, mozkových i periferních arterií, která může vést ke kardiovaskulárnímu onemocnění. U heterozygotů nastávají velké komplikace již kolem 40. roku, které mohou končit úmrtím. U homozygotů může vést k úmrtí jedince již kolem 20. roku.
Další projevy mohou být mnohočetné xantomy, eruptivní xanthomy kůže nebo šlach, arcus corneae et myringis (intersticiální steatóza rohovky nebo bubínku).
Diagnostika steatózy
- Laboratorní testy – sérové transaminázy, feritin.
- Ultrazvuk – hyperechogenní tkáň, obtížná diagnostika u obézních lidí.
- Biopsie – třeba dbát zvýšené opatrnosti (tkáně se steatózou jsou křehké – obzvlášť játra).
Lipidózy

Lipidózy jsou vrozené poruchy enzymů (enzymopatie) lipidového metabolismu. Jde především o lysozomální hydrolázy, které působí rozkládání složených lipidů – vyznačuje se hromaděním (střádáním, thesaurací) lipidů v lysozomálním aparátu. Odbourávání sfingolipidových glykokonjugátů probíhá v lysozomech postupným odštěpováním cukerných jednotek z neredukujícího konce řetězce specifickými exohydrolázami až na ceramid. Podobně sfingomyelin je degradován odštěpením fosforylcholinu. Ceramid je dále deacylován na sfingosin. Tyto konečné produkty opouští lysozom a jsou použity znovu k biosyntéze, nebo jsou dále degradovány. Estery cholesterolu jsou hydrolyzovány, cholesterol je transportován do cytozolu a esterifikován.
Lipidózy bývají kvůli postižení nervového systému někdy označovány také jako neurolipidózy.

Mikroskopie
Hypertrofie lysozomů – mikrovakuolární, pěnitý až voštinovitý vzhled buněk. Následně regresivní změny včetně sekundární tvorby lipopigmentů (ceroid i lipofuscin). Střádanými lipidy bývají gangliosidy, cerebrosidy, sfingomyelin, ceramid, cholesterol a jeho estery. Postihují především histiocyty RES, ale i epitelie a endotelie (viscerální lipidózy) nebo gangliové buňky (neuronální lipidózy).
Rozdělení
- Podle místa postižení
- Neuronální;
- viscerální;
- neuroviscerální;
- podle střádaného lipidu (a defektního enzymu).
Lysozomální onemocnění CNS mají dvě formy:
- postižení gangliových buněk – thesaurující onemocnění;
- postižení bílé hmoty – leukodystrofie (poruchy metabolismu myelinu).
Zjednodušené rozdělení složených lipidů
- Fosfolipidy:
- glycerofosfolipidy – kyselina fosfatidová (3-fosfo-1,2-diacylglyerol) + další složka (cholin, ethanolamin);
- sfingofosfolipidy – ceramid (sfingosin + MK) + fosfát + další složka (je-li jí cholin, jde o sfingomyelin).
- Glykolipidy – obsahují ceramid (sfingosin + MK) s navázanou cukernou složkou:
- cerebrosidy – vazba hexosy (Glc, Gal) na ceramid;
- gangliosidy – vazba oligosacharidu s kys. sialovou (N-acetylneuraminovou) na ceramid.
Gaucherova choroba
- Defekt: deficit glukocerebrosidázy způsobuje hromadění glukocerebrosidů ve slezině (RES) a CNS.
- Klinické příznaky:
- typ 1:
- počátek onemocnění je v dětství, plná manifestace v dospělosti
- typická je splenomegalie, hepatomegalie je jen mírná, ale je možný vznik cirhózy
- dochází k infiltraci kostní dřeně, patologickým frakturám a aseptickým nekrózám
- masivní postižení plic může vést až ke cor pulmonale; známa je též kožní hyperpigmentace a koincidence s různými malignitami
- typ 2:
- mezi základní znaky patří hepatosplenomegalie a těžká neurologická symptomatologie (trismus, strabismus, retroflexe hlavy, progresivní spasticita, hyperreflexie a vznik patologických reflexů, v terminálním stadiu
 hypotonie)
hypotonie)
- mezi základní znaky patří hepatosplenomegalie a těžká neurologická symptomatologie (trismus, strabismus, retroflexe hlavy, progresivní spasticita, hyperreflexie a vznik patologických reflexů, v terminálním stadiu
- typ 3:
- delší průběh nemoci a neuroviscerální symptomatologie kolem 1 - 3 let života, hepatosplenomegalie a později neurologická symptomatologie - ataxie a spastické parézy, poruchy oční motility, mentální retardace a záchvaty (často myoklonie)
- typ 1:
- Mikroskopie: charakteristickým nálezem jsou tzv. Gaucherovy buňky – velké makrofágy střádající lipidy, s „pomačkanou” cytoplasmou, nejprve se objevují v kostní dřeni, později i jinde (podobné buňky, tzv. gaucheroidní, se vyskytují v kostní dřeni při CML)
- Diagnóza: je potvrzena stanovením deficitu aktivity b-glukosidázy v leukocytech izolovaných z periferní krve, nebo kultivovaných kožních fibroblastech; doplňujícím vyšetřením u případů s potvrzenou diagnózou je analýza DNA
- Prenatální diagnostika: v rodinách s enzymaticky prokázanou diagnózou je možná analýzou nativních a kultivovaných choriových klků nebo kultivovaných amniocytů
- Léčba: i.v. dodáním chybějícího enzymu, inhibice biosyntézy glukocerebrosidu
Farberova choroba
- jedná se o AR onemocnění
- Defekt: deficit aktivity kyselé ceramidázy
- Klinické příznaky: poškození podkoží a sliznic deformujícími uzly podmíněnými granulomatosním jizvícím procesem - maximum změn je na kloubech a v okolí šlachových pochev
- postižení hrtanu vede k chraptivosti až k afonii
- bylo popsáno též postižení srdečních chlopní, mírná hepatosplenomegalie, retinální změny podobné tzv. "třešňové skvrně"
- neurologické postižení je méně časté - hypotonie, denervační atrofie a myopatické změny
- mezi základní znaky forem s pozdním nástupem patří mitigované postižení s protrahovaným průběhem (klinicky podobné klasické Farberově chorobě)
- Diagnóza: je potvrzena stanovením deficitu aktivity kyselé ceramidázy v leukocytech izolovaných z periferní krve, nebo kultivovaných kožních fibroblastech
- Prenatální diagnostika: v rodinách s enzymaticky prokázanou diagnosou je možná analýzou nativních a kultivovaných choriových klků nebo kultivovaných amniocytů
- Léčba: není dostupná
Niemann-Pickova choroba
Autosomálně recesivní dědičná střádavá porucha, patří mezi tzv. lipidózy – metabolické poruchy lipidů. Vzniká na podkladě ukládání sfingomyelinu v makrofázích retikuloendotelového systému – převážně v játrech, slezině a kostní dřeni.
Jedná se o heterogenní skupinu onemocnění typu A, B, C, které se liší metabolickou poruchou – deficit kyselé sfingomyelinázy (typ A, B) vs. porucha transportu lipidů (typ C).
Akutní formy typické pro dětský věk postihují nervový systém, chronické se projevují později cholestatickým postižením jater, přecházejícím až do cirhózy. Sekundárně dochází ke zvýšení koncentrací neesterifikovaného cholesterolu.
Niemann-Pickova choroba, typ A a B: deficit aktivity kyselé sfingomyelinázy (je následkem mutace genu SMPD1, je známo více než 100 mutací)
- typ A – mezi základní znaky patří neuroviscerální postižení s úmrtím do 1-3 let věku (specifiky zvýšený výskyt u etnické skupiny aškenázských židů)
- potíže se objevují již v prvních týdnech života
- projevuje se zvracením, průjmy a celkovým neprospíváním novorozence až kachexií; během pár měsíců progreduje v lymfadenopatii a hepatosplenomegalii (vzácně v cholestatický ikterus)
- objevuje se svalová slabost, hypotonie, psychomotorická retardace, postupně dochází ke ztrátě motorických funkcí, spasticitě a rigiditě svalstva; na kůži se mohou vyskytovat xantomy hnědožluté skvrny
- asi u poloviny pacientů se na sítnici objevuje tzv. třešňová skvrna
- pacienti většinou umírají do věku 3 let
- typ B – chronické onemocnění (častěji v jižní Evropě a severní Africe), může se projevit kdykoliv od pozdního dětského věku až do dospělosti
- většinou se projevuje splenomegalií nebo hepatosplenomegalií (těžší onemocnění jater je vzácné)
- často dochází k retikulonodulární RTG infiltraci plic související s intersticiálním postižením, které se může projevit v různé míře námahovou dušností
- dochází také ke zpomalení růstu, opoždění kostní věku a puberty
- intelekt a nervový systém nebývá postižen
- dospělí mívají patologický profil lipidů, trombocytopenii a zvýšenou aktivitu jaterních transamináz
- existují různě závažné formy choroby, většinou s normální délkou života
- Diagnóza Niemann-Pickovy choroby typu A a B: je potvrzena stanovením deficitu aktivity kyselé sfingomyelinázy v leukocytech izolovaných z periferní krve, nebo kultivovaných kožních fibroblastech; doplňujícím vyšetřením u případů s potvrzenou diagnózou je analýza DNA
- Prenatální diagnostika: v rodinách s enzymaticky prokázanou diagnózou je možná analýzou nativních a kultivovaných choriových klků nebo kultivovaných amniocytů; doplňujícím vyšetřením je analýza ultrastruktury choriových klků
- Léčba: terapie rekombinantním enzymem se připravuje
Krabbeho choroba (leukodystrofie)
- Defekt: deficit aktivity galaktocerebrosid b-galaktosidázy
- Klinické příznaky: mezi základní znaky patří manifestace po půl roce života a rychlý průběh
- nejprve je zvýšená iritabilita, hyperestézie, hyperakuzie a zvýšená fotosenzitivita, postupně nastupuje psychomotorická retardace, hypertonie a tonické a klonické záchvaty
- ve finálním stadiu je decerebrace, opistotonus, slepota, popř. hluchota
- exitus nastává okolo 2 let
- laboratorně je nález zvýšené hladiny proteinu v likvoru (zejména albuminu a alfa-2-globulinu) při normálním počtu buněk, atrofie optiku a známky periferní neuropatie (snížena rychlost vedení periferním nervem); EEG může být abnormální, často s fokálními epileptickými záchvaty; na CT a NMR je difuzní atrofie bílé hmoty mozku
- u forem s pozdním nástupem klinických příznaků patří mezi základní znaky - mentální retardace, pyramidové poruchy, poruchy reakce, porucha zraku
- protein v likvoru nemusí být zvýšený, rychlost vedení periferním nervem může být normální, nebo snížena
- Diagnóza: je potvrzena stanovením deficitu aktivity galaktocerebrosid-b-galatozidázy v leukocytech izolovaných z periferní krve, nebo kultivovaných kožních fibroblastech
- Prenatální diagnostika: v rodinách s enzymaticky prokázanou diagnózou je možná analýzou nativních a kultivovaných choriových klků nebo kultivovaných amniocytů
- Léčba: není dostupná
Metachromatická leukodystrofie
- Defekt: deficit aktivity arylsulfatázy A
- Klinické příznaky: mezi základní znaky patří poruchy chůze, mentální regrese, ataxie, ztráta řeči, periferní neuropatie, kvadruparéza, atrofie očního nervu, šedavé zbarvení makuly
- choroba trvá několik měsíců
- laboratorně je nález zvýšené hladiny proteinu v likvoru (zejména albuminu a alfa-2-globulinu) při normálním počtu buněk, atrofie optiku a známky periferní neuropatie (snížena rychlost vedení periferním nervem); EEG může být abnormální, často s fokálními epileptickými záchvaty; na CT a NMR je difuzní atrofie bílé hmoty mozku
- u forem s pozdním nástupem klinických příznaků patří mezi základní znaky mentální retardace, psychotické příznaky, pyramidové poruchy, poruchy reakce, porucha zraku
- protein v likvoru nemusí být zvýšený, rychlost vedení periferním nervem může být normální, nebo snížená
- v moči je mnohonásobně zvýšená koncentrace sulfatidu
- Diagnóza: je potvrzena stanovením deficitu aktivity arylsulfatázy A v leukocytech izolovaných z periferní krve, nebo kultivovaných kožních fibroblastech; doplňujícím vyšetřením u případů s potvrzenou diagnózou je analýza DNA
- Prenatální diagnostika: v rodinách s enzymaticky prokázanou diagnózou je možná analýzou nativních a kultivovaných choriových klků nebo kultivovaných amniocytů
- Léčba: není dostupná
Tay-Sachsova choroba (GM2 gangliosidóza)
- Defekt: deficit aktivity N-acetyl-beta-D-glukózaminidázy A
- Klinické příznaky: existují klinické varianty podle doby nástupu choroby a závažnosti projevu
- u infantilní formy patří mezi základní znaky progredující neurologická symptomatologie, hypotonie, myoklonie, křeče, dále třešňová skvrna na očním pozadí, progresivní psychomotorická deteriorace, makrocefalie, a exitus do 2-4 let; frekvence výskytu choroby je vysoká u aškenázských Židů
- u infantilního typu s pozdějším nástupem patří k základním symptomům centrální neurologická symptomatologie a tezaurizační retinopatie
- neurologické postižení je velmi variabilní - může dominovat klasické postižení CNS (dystonie, extrapyramidové příznaky, ataxie), ale může být i obraz juvenilní spinální svalové atrofie (typu Kugelberga-Walanderové), systémové atrofie blízké amyotrofické laterální skleróze nebo progresivní spinocerebelární ataxie Friedreichova typu
- typické je hromadění GM2 gangliosidu v mozku
- Diagnóza: je potvrzena stanovením deficitu aktivity N-acetyl-beta-D-glukózaminidázy A v leukocytech izolovaných z periferní krve, nebo kultivovaných kožních fibroblastech
- Prenatální diagnostika: v rodinách s enzymaticky prokázanou diagnózou je možná analýzou nativních a kultivovaných choriových klků nebo kultivovaných amniocytů
- Léčba: není dostupná
Fabryho choroba
- jedná se o X vázané onemocnění, frekvence 1:40 000
- Defekt: deficit aktivity alfa-galaktozidázy A
- Klinické příznaky: mezi základní znaky patří u hemizygotů (mužů) trvalé nebo epizodické akroparestézie nebo palčivá bolest různé intenzity, mírně zvýšená teplota a sedimentace
- charakteristické jsou výsevy kožních angiokeratomů, zákal rohovky a deformity retinálních a spojivkových cév
- renální postižení zahrnuje lipidurii, proteinurii a progredující insuficienci
- kardiovaskulární postižení zahrnuje hypertenzi (renální), hypertrofii myokardu (kardiomegalie) a ischemické změny různých orgánů, zejména mozku
- může být přítomna centrální neurologická symptomatologie
- u heterozygotů (žen) je postižení různé - plně vyvinuté příznaky až jejich úplná absence
- v moči je mnohonásobně zvýšená koncentrace globotriaosylceramidu
- Diagnóza: je potvrzena stanovením deficitu aktivity a-galaktozidázy A v leukocytech izolovaných z periferní krve, nebo v kultivovaných kožních fibroblastech; doplňujícím vyšetřením u případů s potvrzenou diagnózou je analýza DNA, pro potvrzení heterozygotního stavu je však nezbytná
- Prenatální diagnostika: v rodinách s enzymaticky prokázanou diagnózou je možná v nativních a kultivovaných choriových klcích nebo kultivovaných amniocytech; doplňujícím vyšetřením je analýza ultrastruktury choriových klků
- Léčba: terapie je možná i.v. dodáním rekombinantní a-galaktozidázy A
